
Behördliche Genehmigungsverfahren für Medizinprodukte in den USA: Der Unterschied zwischen 510(k) und PMA
Inhaltsverzeichnis
- Was ist 510(k)?
- Was ist die Vorabgenehmigung (PMA)?
- Warum müssen wir Zulassungsverfahren wie 510(k) oder PMA durchlaufen?
- Wichtige FDA-Dokumente
- Wann ist eine neue 510(k) für eine Änderung eines bestehenden Produkts einzureichen?
- Zusammenfassung
Planen Sie, ein medizinisches Gerät in den US-Markt einzuführen? Dann ist es wichtig, dass Sie die Vorschriften der US-amerikanischen Lebens- und Arzneimittelbehörde (Food and Drug Administration, FDA) kennen.
Bevor Sie Ihr Produkt in den USA verkaufen können, muss es zunächst von der FDA zugelassen werden. Dafür gibt es zwei Möglichkeiten - das 510(k)-Verfahren und die Vorabgenehmigung (Pre-Market Approval, PMA). Beide Verfahren haben ihre eigenen Schritte und Anforderungen. Welches Verfahren das richtige ist, hängt vor allem von der Art des Produkts ab, das Sie herstellen, und davon, wie riskant es für Patienten oder Anwender sein könnte.
In diesem Artikel geben wir Ihnen einen klaren Überblick über beide Optionen, erklären, wie sie sich unterscheiden, und helfen Ihnen dabei, zu verstehen, wann sie jeweils eingesetzt werden - und was das Verfahren beinhaltet.
Was ist 510(k)?
510(k), offiziell Premarket Notification genannt, ist ein Zulassungsverfahren, das für Medizinprodukte mit geringem bis mittlerem Risiko (Klasse I und II) vorgesehen ist. In diesem Verfahren muss der Hersteller nachweisen, dass das neue Produkt im Wesentlichen gleichwertig mit einem bereits zugelassenen Produkt (dem sogenannten Prädikatsprodukt) ist.
Wann wird es angewendet?
- Bei der Einführung eines neuen Produkts auf dem Markt
- Bei einer wesentlichen Änderung eines bestehenden Produkts (z. B. seiner Technologie, seines Materials oder seiner Software)
Was ist die Vorabgenehmigung (PMA)?
PMA - Pre-Market Approval - ist ein Verfahren, das für Hochrisikoprodukte (Klasse III) wie Herzschrittmacher, Kunstherzen oder Implantate vorgesehen ist. Dieses Verfahren ist anspruchsvoller und erfordert klinische Studien, Labortests und Nachweise für die Sicherheit und Wirksamkeit des Produkts.
Wann wird es angewendet?
- Wenn es noch kein ähnliches Produkt (Prädikat) auf dem Markt gibt
- Wenn das Produkt eine neue Technologie beinhaltet oder ein höheres Risiko darstellt
Warum müssen wir Zulassungsverfahren wie 510(k) oder PMA durchlaufen?
Jedes Medizinprodukt, das in den USA auf den Markt gebracht werden soll, muss von der FDA (U.S. Food and Drug Administration) zugelassen werden. Ohne diese Zulassung kann das Produkt in den USA nicht legal verkauft oder vermarktet werden.
Das Genehmigungsverfahren soll Folgendes sicherstellen:
- Sicherheit und Wirksamkeit - Überprüfung, ob das Produkt den Patienten nicht schadet und wie vorgesehen funktioniert
- Sicherung der Qualität
- Patientenschutz - Sicherstellung, dass das Produkt keine neuen, unerwarteten Risiken birgt.
Was ist eine wesentliche Änderung und welche Rolle spielt sie im
Genehmigungsverfahren?
Sobald ein Produkt zugelassen ist (z. B. durch das 510(k)-Verfahren) und der Hersteller eine Änderung vornehmen möchte, muss er prüfen, ob die Änderung “wesentlich” ist.
- Ist die Änderung nicht wesentlich, kann das Produkt weiterhin unter der ursprünglichen Zulassung vermarktet werden.
- Handelt es sich um eine wesentliche Änderung, ist ein neuer Antrag erforderlich - beispielsweise eine neue 510(k)-Meldung -, da die Änderung die Sicherheit, Wirksamkeit oder Zweckbestimmung des Produkts beeinträchtigen könnte.
Beispiel dafür, was eine wesentliche Änderung ist und was nicht:
Wenn Sie einen Gefäßkatheter haben und nur seine Farbe ändern, ist dies in der Regel keine wesentliche Änderung. Wenn Sie jedoch das Material ändern, das mit Blut in Berührung kommt, wird dies als wesentliche Änderung betrachtet, da dies die Biokompatibilität und Sicherheit des Produkts beeinträchtigen könnte.
Wichtige FDA-Dokumente
Im Rahmen des FDA-Zulassungsverfahrens legt die Behörde klare Regeln für das weitere Vorgehen auf der Grundlage der Risikoklassifizierung des Medizinprodukts fest. Um die Hersteller bei der Navigation durch das Verfahren zu unterstützen, stellt die FDA offizielle Leitfäden zur Verfügung, in denen detailliert erläutert wird, wie der richtige Zulassungsweg zu wählen ist und welche Begleitdokumente erforderlich sind.
Nachstehend finden Sie Links zu den beiden wichtigsten Dokumenten, die jeder Hersteller von Medizinprodukten kennen sollte:
1. Entscheidung über die Einreichung eines 510(k)-Antrags für eine Änderung
an einem bestehenden Produkt
https://www.fda.gov/media/99812/download
Was beinhaltet dieses Dokument? Dieser Leitfaden richtet sich an Hersteller, die bereits über ein von der FDA zugelassenes Produkt (über den 510(k)-Zulassungsweg) verfügen und Änderungen daran vornehmen möchten. Die FDA erläutert ausführlich, wie zu beurteilen ist, ob die Änderung wesentlich genug ist, dass eine neue 510(k)-Anmeldung erforderlich ist.
2. Vorabgenehmigung (PMA)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma
Was beinhaltet dieses Dokument? Diese Webseite beschreibt den gesamten Prozess der PMA (Pre-Market Approval) - das strengste FDA-Zulassungsverfahren, das für Hochrisikoprodukte (Klasse III) erforderlich ist.
Wann ist eine neue 510(k) für eine Änderung eines bestehenden Produkts
einzureichen?
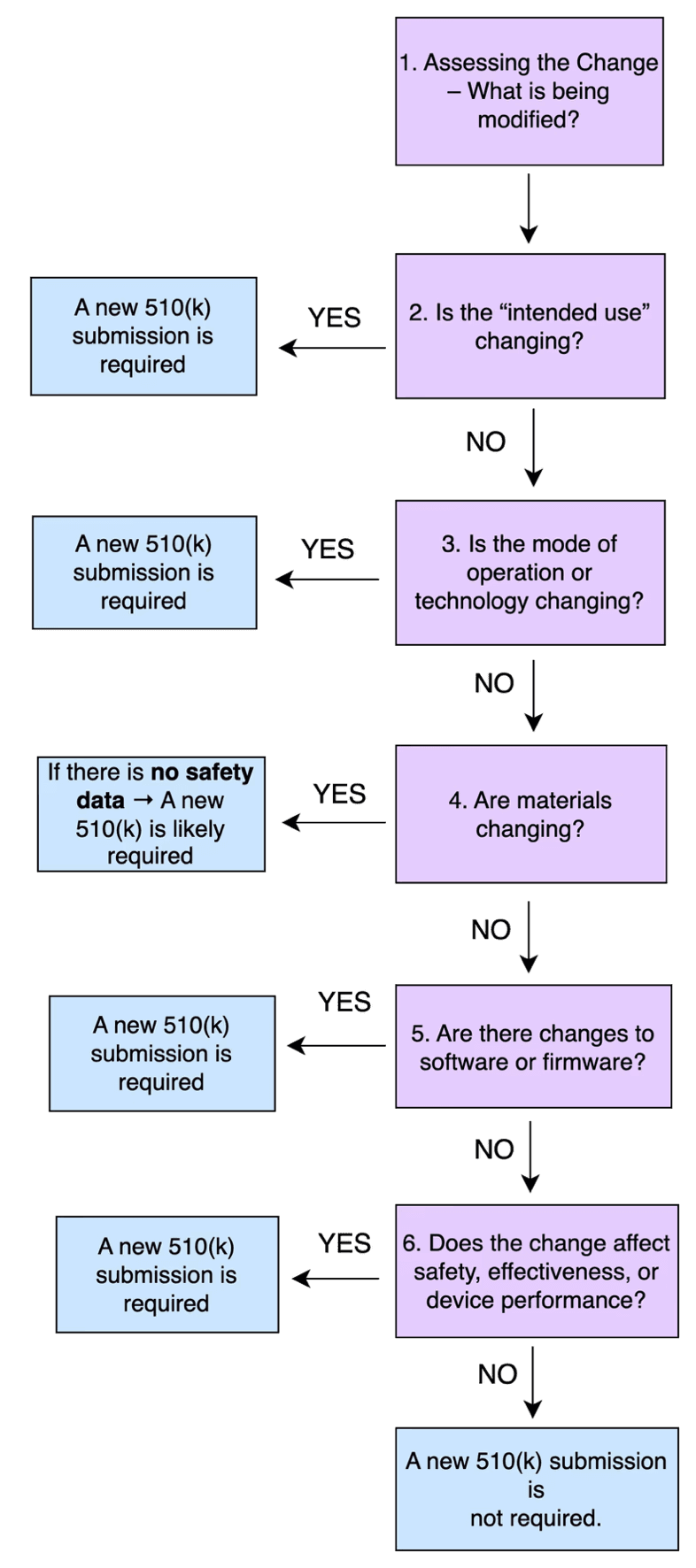
- Wenn die Änderung wesentlich ist → müssen Sie eine neue 510(k) vorbereiten und bei der FDA einreichen
- Wenn die Änderung unwesentlich ist → genügt es, die Änderung intern zu dokumentieren und die Aufzeichnungen in Ihrem Qualitätsmanagementsystem (QMS) aufzubewahren
- Wenn Sie sich unsicher sind → sollten Sie einen Experten für Zulassungsfragen konsultieren oder ein Vorabgespräch mit der FDA vereinbaren
Zusammenfassung
Wenn Sie planen, ein Medizinprodukt auf den US-Markt zu bringen, ist es entscheidend, korrekt zu bestimmen, ob das 510(k)-Verfahren ausreicht oder ob das anspruchsvollere Pre-Market Approval-Verfahren (PMA) erforderlich ist. Jeder dieser Zulassungswege verfügt über klar definierte Regeln und Kriterien.
Die Wahl des falschen Verfahrens kann dazu führen, dass die FDA Ihren Antrag ablehnt, was sich erheblich auf Ihren Projektzeitplan und die Markteinführung des Produkts auswirken kann.
Wie bereits erwähnt, dauert die 510(k)-Freigabe in der Regel 90 bis 180 Tage. Das PMA-Verfahren ist wesentlich länger und kann je nach Komplexität des Produkts und dem Bedarf an klinischen Daten mehrere Monate bis über ein Jahr dauern.
Lehnt die FDA den Antrag aufgrund eines falsch gewählten Verfahrens oder unzureichender Unterlagen ab, kann es notwendig sein, das Verfahren neu zu beginnen.
Fehler dieser Art können zu folgenden Konsequenzen führen:
- Verzögerungen von mehreren Monaten bis zu einem Jahr
- erhöhte Kosten für die Erstellung neuer Unterlagen
- oder sogar zum Verlust von Marktchancen für das Produkt
Aus diesem Grund empfehlen wir dringend die Zusammenarbeit mit Experten für die Zulassung von Medizinprodukten, die Ihnen dabei helfen können, die Risikoklasse des Produkts einzuschätzen, die geeignete Strategie zu wählen und sicherzustellen, dass die FDA-Anträge auf Anhieb korrekt und vollständig sind.