
Klassifizierung von Medizinprodukten nach MDR: Was Sie wissen müssen
Inhaltsverzeichnis
- Was ist die MDR?
- Wie werden Medizinprodukte gemäß der MDR klassifiziert und in den Markt eingeführt?
- Was ist eine benannte Stelle?
- Was ist Post-Market Surveillance (PMS)?
- Fazit und Empfehlungen
- Benötigen Sie Unterstützung bei der EU MDR 2017/745? Wir helfen Ihnen gerne.
Wenn Sie Medizinprodukte in der EU herstellen oder verkaufen, müssen Sie die Vorschriften der europäischen Medizinprodukteverordnung (MDR) einhalten. Diese Verordnung gilt seit Mai 2021 und zielt in erster Linie darauf ab, die Sicherheit, Zuverlässigkeit und Rückverfolgbarkeit von Medizinprodukten zu gewährleisten.
Ein Medizinprodukt kann alles Mögliche sein – von einem Pflaster über eine Injektionsnadel, einen Herzschrittmacher bis hin zu medizinischer Software. Jedes Produkt unterscheidet sich jedoch in seiner Komplexität und seiner Wechselwirkung mit dem menschlichen Körper, und genau das bestimmt seine Einstufung in eine von vier Risikoklassen.
Was ist die MDR?
Die MDR (Medizinprodukteverordnung, Medical Device Regulation) ist die europäische Verordnung Nr. 2017/745, die 2021 in Kraft trat. Ihr Ziel ist es, die Sicherheit und Rückverfolgbarkeit von Medizinprodukten auf dem EU-Markt zu erhöhen.
Die MDR legt besonderen Wert auf:
- Strengere klinische Bewertungen und Überwachung nach dem Inverkehrbringen
- Einführung des UDI-Systems (Unique Device Identifier)
- Verpflichtende Registrierung von Produkten und Herstellern im EUDAMED-System
- Sorgfältige Klassifizierung der Produkte nach ihrem Risikoniveau
Wie werden Medizinprodukte gemäß der MDR klassifiziert und in den Markt eingeführt?
Klasse I – Produkte mit dem geringsten Risiko
Diese Kategorie umfasst einfache Produkte, die nur auf der Körperoberfläche angewendet werden und den Körper nicht durchdringen.
Beispiele: Pflaster, Verbände, Einweghandschuhe, Dentalspiegel, manuelle Rollstühle
Was sind die Anforderungen für Medizinprodukte der Klasse I?
Der Hersteller führt die Konformitätsbewertung selbst durch.
- Erstellung der vollständigen technischen Dokumentation (gemäß Anhang II und III der MDR)
- Durchführung einer klinischen Bewertung
- Implementierung eines Systems zur Überwachung nach dem Inverkehrbringen (Post-Market-Surveillance)
- Registrierung des Produkts in der EUDAMED-Datenbank
- Vergabe einer eindeutigen Produktkennzeichnung (UDI)
- Anbringen der CE-Kennzeichnung
Falls das Produkt jedoch steril ist, eine Messfunktion enthält (z. B. Thermometer) oder ein wiederverwendbares chirurgisches Instrument darstellt, muss eine benannte Stelle (eine von der EU zugelassene unabhängige Organisation) für die Bewertung dieser spezifischen Eigenschaften hinzugezogen werden.
Unterkategorien der Klasse I:
- Is – sterile Produkte
- Im – Produkte mit Messfunktion
- Ir – wiederverwendbare chirurgische Instrumente
Klasse IIa – Produkte mit mittlerem Risiko
Hierbei handelt es sich um Produkte, die kurzzeitig in den Körper gelangen oder in begrenztem Maße mit ihm interagieren.
Beispiele: Kontaktlinsen, Injektionsnadeln, Ultraschall-Diagnosegeräte, medizinische Software mit Entscheidungshilfen
Was sind die Anforderungen für Medizinprodukte der Klasse IIa??
-
Der Hersteller arbeitet mit einer benannten Stelle zusammen, die die technische Dokumentation, Produktsicherheit und das Qualitätsmanagementsystem des Herstellers überprüft.
-
Zusätzlich bereitet der Hersteller eine klinische Bewertung, einen Plan zur Überwachung nach dem Inverkehrbringen, die UDI-Vergabe und die Registrierung im EUDAMED-System vor.
Klasse IIb – Produkte mit höherem Risiko
Diese Produkte werden über einen längeren Zeitraum verwendet oder beeinflussen lebenswichtige Körperfunktionen.
Beispiele: Infusionspumpen, Beatmungsgeräte, Röntgengeräte, spezielle Krankenhausbetten
Was sind die Anforderungen für Medizinprodukte der Klasse IIb?
- Die Beteiligung einer benannten Stelle ist obligatorisch
- Der Prozess ist komplexer mit stärkerem Fokus auf Sicherheit, technische Dokumentation und klinische Daten
- Der Hersteller reicht die technische Dokumentation zur Prüfung ein und kann nach Freigabe CE-Kennzeichnung, EUDAMED-Registrierung und Markteinführung vornehmen
- Vollständiges Qualitätsmanagementsystem erforderlich
- Klinische Studien sind häufig erforderlich, diese sind jedoch nicht automatisch vorgeschrieben wie bei Klasse III. Die Notwendigkeit hängt vom Produkttyp, klinischen Daten und Äquivalenznachweis ab
Klasse III – Produkte mit höchstem Risiko
Dazu gehören Geräte, die in den Körper implantiert werden, lebenserhaltend sind oder einen direkten und wesentlichen Einfluss auf die Gesundheit eines Patienten haben.
Beispiele: Herzschrittmacher, künstliche Herzklappen, Stents, Hirnelektroden
Was sind die Anforderungen für Medizinprodukte der Klasse III?
Dies ist der anspruchsvollste Prozess. Die benannte Stelle muss alle Aspekte des Produkts gründlich überprüfen – von der technischen Dokumentation über klinische Daten und Herstellungsverfahren bis hin zur Sicherheit.
Die Inverkehrbringung eines Klasse-III-Produkts erfordert:
- Detaillierte technische Dokumentation – eine benannte Stelle führt eine umfassende Prüfung durch; erst nach deren Freigabe dürfen CE-Kennzeichnung, EUDAMED-Registrierung und Markteinführung erfolgen
- Klinische Studie: Verpflichtend. Der Hersteller muss Daten aus einer eigenen klinischen Studie vorlegen, die die Sicherheit, Wirksamkeit und den klinischen Nutzen des Produkts für den Patienten nachweisen
- Ein voll funktionsfähiges Qualitätsmanagementsystem
- Ein wirksamer Plan zur Überwachung nach dem Inverkehrbringen
- Ein Plan zur klinischen Nachbeobachtung nach dem Inverkehrbringen (fortlaufende klinische Überwachung nach Markteinführung des Produkts)
| Klasse | Benannte Stelle | Klinische Bewertung | Klinische Studie | QMS | Klinische Nachbeobachtung | EUDAMED / UDI / CE |
|---|---|---|---|---|---|---|
| I | ❌ Nein ✅(Ausnahmen Is, Im, Ir – nur für bestimmte Aspekte) | ✅ Ja | ❌ Nein | ❌ Nein | ⚠️ Basis-PMS | ✅ Ja |
| IIa | ✅ Ja | ✅ Ja | ⚠️ Möglich | ✅ Ja | ⚠️ Manchmal | ✅ Ja |
| IIb | ✅ Ja | ✅ Intensiv | ✅ Oft | ✅ Ja | ✅ Ja | ✅ Ja |
| III | ✅ Ja | ✅ Sehr detailliert | ✅ Fast immer | ✅ Ja | ✅ Ja | ✅ Ja |
Was ist eine benannte Stelle?
Eine benannte Stelle ist eine unabhängige Organisation (meist ein spezialisiertes Unternehmen oder Institut), die von der EU offiziell ernannt wurde, um zu prüfen, ob ein Medizinprodukt alle Anforderungen der MDR erfüllt.
Ihre Aufgabe besteht darin, die Produktdokumentation, die Sicherheit, die Fertigungsqualität und gegebenenfalls die klinischen Daten des Produkts zu überprüfen.
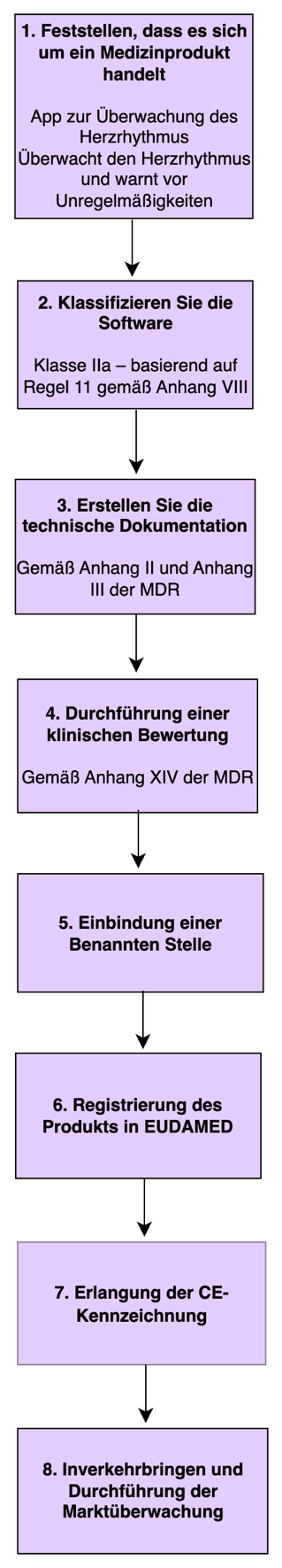
Schritt-für-Schritt-Prozess zur Zulassung von medizinischer Software (Klasse IIa – Herzrhythmus-Überwachungs-App)
Was ist Post-Market Surveillance (PMS)?
Die PMS (Überwachung nach dem Inverkehrbringen) ist ein verpflichtender Prozess, bei dem der Hersteller die Leistung, Sicherheit und Qualität eines Medizinprodukts nach Markteinführung und Anwendung im Alltag überwacht.
Die Ziele der Post-Market Surveillance (PMS) sind:
- Erkennung potenzieller Nebenwirkungen, Fehler oder Funktionsstörungen
- Sicherstellung, dass das Produkt weiterhin wie vorgesehen funktioniert
- Einholung von Nutzerfeedback (z. B. von Ärzten, Patienten)
- Unterstützung der kontinuierlichen Produktverbesserung
Was bedeutet Post-Market Surveillance (PMS) für Hersteller?
Jeder Hersteller eines Medizinprodukts unter der europäischen MDR muss ein PMS-System einrichten, unabhängig von der Risikoklasse. Umfang und Form der Verpflichtungen variieren jedoch je nach Klassifizierung.
Zu diesem Zweck werden zwei Hauptdokumente verwendet:
PMS-Bericht (für Klasse I)
- Internes Dokument, muss nicht regelmäßig bei benannter Stelle eingereicht werden
- Es muss jedoch jederzeit verfügbar und auf dem neuesten Stand sein, insbesondere für Inspektionen oder Audits
PSUR – Periodischer Sicherheitsbericht (für die Klassen IIa, IIb, III)
- Formeller Bericht, der regelmäßig aktualisiert und ggf. bei benannter Stelle eingereicht werden muss (sofern eine solche beteiligt ist)
- Enthält Informationen zu Sicherheit, Leistung, PMS-Ergebnissen und etwaigen Korrekturmaßnahmen
Übersicht PMS-Berichtspflichten nach Risikoklasse
| Klasse | Berichtstyp | Häufigkeit |
|---|---|---|
| I | PMS-Bericht | Nach Bedarf (nicht streng definiert) |
| IIa | PSUR | Mindestens alle 2 Jahre |
| IIb | PSUR | Jährlich |
| III | PSUR | Jährlich |
Fazit und Empfehlungen
Die MDR stellt strengere Anforderungen, hilft aber, qualitativ hochwertige, funktionale und sichere Produkte von unzuverlässigen Lösungen zu unterscheiden. Für seriöse Softwarehersteller ist das eine Chance, Vertrauen bei Fachkräften und Patienten zu gewinnen und langfristige Wettbewerbsfähigkeit auf dem Markt zu gewährleisten. Der Prozess der Zulassung eines Medizinprodukts gemäß der europäischen MDR-Verordnung ist anspruchsvoll, aber essenziell für die Patientensicherheit und das Vertrauen in das Produkt. Trotz der Komplexität ist er klar strukturiert und bietet Raum für Innovationen, wenn der Hersteller systematisch und qualitätsorientiert vorgeht.
Wenn Sie über kein internes Team für die regulatorische Compliance verfügen, kann die Zusammenarbeit mit einem qualifizierten Beratungsunternehmen den Zertifizierungsprozess erheblich beschleunigen und das Risiko einer Ablehnung oder von Komplikationen während Audits verringern. Klinische Experten und Qualitätsmanagementberater helfen Ihnen, Fehler zu vermeiden, die sonst zu Verzögerungen oder zum Scheitern der Zertifizierung führen könnten.
MDR kann eine Herausforderung sein, aber mit der richtigen Herangehensweise und erfahrenen Partnern lässt sich auch komplexe medizinische Software sicher, effizient und vertrauenswürdig auf den Markt bringen.
Benötigen Sie Unterstützung bei der EU MDR 2017/745? Wir helfen Ihnen gerne.
Wir haben bereits zahlreichen Medizinprodukten erfolgreich zum Marktzugang verholfen – und sind bereit, auch Ihr Produkt zu unterstützen. Unser Team hat an groß angelegten regulatorischen und entwicklungsbezogenen Projekten mit internationalen Marktführern wie Merck, Ypsomed, Novartis und Dentsply Sirona zusammengearbeitet. Ob es um die Klassifizierung, die Erstellung der technischen Dokumentation oder den Aufbau eines konformen Qualitätsmanagementsystems (QMS) geht – wir bringen praktische Erfahrung und fundiertes regulatorisches Wissen in jeden Schritt des Prozesses ein.